
YERVOY 5 mg-ml, solution à diluer pour perfusion, boîte de 1 flacon de 10 ml
Retiré du marché le : 26/11/2021
Dernière révision : 02/04/2021
Taux de TVA : 0%
Laboratoire exploitant : BRISTOL-MYERS SQUIBB
Source :

Mésothéliome Pleural Malin
YERVOY est indiqué en association au nivolumab, en première ligne, dans le traitement des patients adultes atteints d'un mésothéliome pleural malin non résécable.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
YERVOY en association à nivolumab
Lorsqu'ipilimumab est administré en association, se référer au Résumé des Caractéristiques du Produit des autres traitements en association avant l'initiation du traitement. Pour des informations complémentaires sur les mises en garde et précautions d'emploi associées au traitement nivolumab, se référer au RCP de nivolumab. La plupart des effets indésirables d'origine immunologique s'est améliorée ou résolue avec une prise en charge appropriée, incluant l'initiation de corticoïdes et des modifications de traitement (voir rubrique Posologie et mode d'administration). Des effets indésirables d'origine immunologique sont survenus à des fréquences plus élevées lorsque le nivolumab a été administré en association à l'ipilimumab comparativement au nivolumab en monothérapie.
Des événements indésirables cardiaques et pulmonaires incluant des embolies pulmonaires ont aussi été rapportés avec le traitement en association. Les patients doivent être surveillés en continu pour des effets indésirables cardiaques et pulmonaires, ainsi que pour des signes cliniques, des symptômes et des anomalies biologiques indiquant des troubles électrolytiques et une déshydratation, avant l'initiation et à intervalles réguliers au cours du traitement. Ipilimumab en association à nivolumab doit être arrêté en cas d'effets indésirables sévères cardiaques et pulmonaires récurrents ou pouvant menacer le pronostic vital (voir rubrique Posologie et mode d'administration).
Les patients doivent être surveillés en continu (au moins jusqu'à 5 mois après la dernière perfusion), un effet indésirable avec ipilimumab en association à nivolumab pouvant survenir à tout moment pendant ou après l'arrêt du traitement.
Effets indésirables d'origine immunologique
Ipilimumab est associé à des effets indésirables inflammatoires résultant d'une réponse immunitaire élevée ou excessive (effets indésirables d'origine immunologique), vraisemblablement liée à son mécanisme d'action. Les effets indésirables d'origine immunologique, qui peuvent être sévères ou menaçant le pronostic vital, peuvent concerner les systèmes gastro-intestinaux, hépatiques, cutanés, nerveux, endocriniens ou d'autres systèmes d'organes. Bien que la plupart des effets indésirables d'origine immunologique apparaissent pendant la période d'induction, leur survenue plusieurs mois après la dernière administration d'ipilimumab a également été rapportée. La diarrhée, augmentation de la fréquence des selles, selles sanglantes, élévation des tests hépatiques, éruption cutanée et endocrinopathie doivent être considérés comme inflammatoires et liés à ipilimumab, sauf si une autre étiologie a été identifiée. Un diagnostic précoce et une prise en charge appropriée sont essentiels pour minimiser les complications menaçant le pronostic vital.
Une corticothérapie systémique à forte dose avec ou sans traitement immunosuppresseur additionnel peut être nécessaire pour la prise en charge des effets indésirables sévères d'origine immunologique. Les recommandations de prise en charge des effets indésirables d'origine immunologique observés avec ipilimumab en association à nivolumab sont décrites ci-dessous:
En cas de suspicion d'effets indésirables d'origine immunologique, une évaluation appropriée doit être effectuée afin de confirmer l'étiologie ou d'exclure d'autres causes. Sur la base de la sévérité de l'effet indésirable, le traitement par ipilimumab en association au nivolumab doit être suspendu, et des corticoïdes administrés. Si une immunosuppression par corticoïdes est utilisée pour traiter un effet indésirable relié à une thérapie combinée, une décroissance progressive des doses sur une période d'au moins un mois doit être initiée à partir de l'amélioration. Une diminution rapide des doses peut entraîner une aggravation ou une récidive de l'effet indésirable. Des traitements immunosuppresseurs non stéroïdiens doivent être ajoutés en cas d'aggravation ou d'absence d'amélioration malgré l'utilisation de corticoïdes.
Le traitement par ipilimumab en association au nivolumab ne doit pas être repris tant que le patient reçoit des doses immunosuppressives de corticoïdes ou d'autres médicaments immunosuppresseurs. Une prophylaxie antibiotique doit être utilisée pour prévenir les infections opportunistes chez les patients recevant des médicaments immunosuppresseurs.
Ipilimumab en association au nivolumab doit être définitivement arrêté en cas d'effet indésirable sévère récurrent d'origine immunologique, et pour tout effet indésirable d'origine immunologique mettant en jeu le pronostic vital.
Colite d'origine immunologique Ipilimumab en association à nivolumab
Des diarrhées ou des colites sévères ont été observées avec ipilimumab en association à nivolumab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour déceler l'apparition de diarrhées et d'autres symptômes de colites, tels que des douleurs abdominales et la présence de mucus ou de sang dans les selles. Une étiologie infectieuse ou liée à la maladie doit être éliminée.
En cas de diarrhée ou de colite de Grade 4, ipilimumab en association à nivolumab doit être arrêté définitivement et une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit être débutée.
La survenue de diarrhée ou colite de Grade 3 avec ipilimumab en association à nivolumab nécessite aussi un arrêt définitif du traitement et l'initiation de corticoïdes à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent.
En cas de diarrhée ou de colite de Grade 2, ipilimumab en association à nivolumab doit être suspendu. Les diarrhées ou les colites persistantes doivent être traitées par une corticothérapie à la dose de 0,5 à 1 mg/kg/jour de méthylprednisolone ou équivalent. Après amélioration, ipilimumab en association à nivolumab peut être repris si nécessaire après réduction progressive des corticoïdes.
En cas d'aggravation ou d'absence d'amélioration malgré l'initiation d'une corticothérapie, les doses de corticoïdes doivent être augmentées à 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent, et ipilimumab en association à nivolumab doit être arrêté définitivement.
Pneumopathie inflammatoire d'origine immunologique
Ipilimumab en association à nivolumab
Des pneumopathies inflammatoires ou interstitielles sévères, dont des cas d'issue fatale, ont été observées avec ipilimumab en association à nivolumab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour déceler l'apparition de signes et symptômes de pneumopathie inflammatoire, tels que des modifications radiologiques (ex : opacités focales en verre dépoli, infiltrats localisés), dyspnée et hypoxie. Une étiologie infectieuse ou liée à la maladie doit être éliminée.
En cas de pneumopathie inflammatoire de Grade 3 ou 4, ipilimumab en association à nivolumab doit être arrêté définitivement et une corticothérapie à la dose de 2 à 4 mg/kg/jour de méthylprednisolone ou équivalent doit être débutée.
En cas de pneumopathie inflammatoire (symptomatique) de Grade 2, ipilimumab en association à nivolumab doit être suspendu et une corticothérapie à la dose de 1 mg/kg/jour de méthylprednisolone ou équivalent doit être débutée. Après amélioration, ipilimumab en association à nivolumab peut être repris après réduction progressive des corticoïdes. En cas d'aggravation ou d'absence d'amélioration malgré l'initiation d'une corticothérapie, les doses de corticoïdes doivent être augmentées à 2 à 4 mg/kg/jour de méthylprednisolone ou équivalent, et ipilimumab en association à nivolumab doit être arrêté définitivement.
Hépatotoxicités d'origine immunologique
Ipilimumab en association à nivolumab
Des hépatites sévères ont été observées avec ipilimumab en association à nivolumab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour déceler l'apparition de signes et symptômes d'hépatite, tels que des augmentations des transaminases et de la bilirubine totale. Une étiologie infectieuse ou liée à la maladie doit être éliminée.
En cas d'élévation de Grade 3 ou 4 des transaminases ou de la bilirubine totale, ipilimumab en association à nivolumab doit être arrêté définitivement et une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit être débutée.
En cas d'élévation de Grade 2 des transaminases ou de la bilirubine totale, ipilimumab en association à nivolumab doit être suspendu. La persistance de cette élévation des valeurs biologiques doit être prise en charge par une corticothérapie à la dose de 0,5 à 1 mg/kg/jour de méthylprednisolone ou équivalent. Après amélioration, ipilimumab en association à nivolumab peut être repris si nécessaire après réduction progressive des corticoïdes. En cas d'aggravation ou d'absence d'amélioration malgré l'initiation d'une corticothérapie, les doses de corticoïdes doivent être augmentées à 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent, et ipilimumab en association à nivolumab doit être arrêté définitivement.
Effets indésirables cutanés d'origine-immunologique
L'utilisation d'ipilimumab en association à nivolumab doit être considérée avec précaution chez un patient ayant présenté un effet indésirable cutané sévère ou ayant menacé le pronostic vital lors d'un précédent traitement anticancéreux stimulant l'immunité.
Ipilimumab en association à nivolumab
Des éruptions cutanées sévères ont été observées avec ipilimumab en association à nivolumab (voir rubrique Effets indésirables). Ipilimumab en association à nivolumab doit être suspendu en cas d'éruption cutanée de Grade 3, et il doit être arrêté définitivement en cas d'éruption cutanée de Grade 4. Les éruptions cutanées sévères doivent être pris en charge avec de hautes doses de corticoïdes, de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent.
De rares cas de SJS et de NET, dont certains d'issue fatale, ont été observés. En cas d'apparition de signes ou symptômes de SJS ou de NET, le traitement par ipilimumab en association à nivolumab doit être interrompu et le patient adressé à un service spécialisé pour évaluation et traitement.
Si le patient a développé un SJS ou une NET lors de l'utilisation d'ipilimumab en association à nivolumab, l'arrêt définitif du traitement est recommandé (voir rubrique Posologie et mode d'administration).
Néphrite et atteinte rénale d'origine immunologique
Ipilimumab en association à nivolumab
Des néphrites et des atteintes rénales sévères ont été observées avec le traitement ipilimumab en association à nivolumab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour déceler l'apparition de signes et symptômes de néphrite ou d'atteinte rénale. La plupart des patients ont présenté des augmentations asymptomatiques de la créatinine sérique. Toute autre étiologie liée à la maladie doit être écartée.
En cas d'élévation de Grade 4 de la créatinine sérique, ipilimumab en association à nivolumab doit être arrêté définitivement et une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit être débutée.
En cas d'élévation de Grade 2 ou 3 de la créatinine sérique, ipilimumab en association à nivolumab doit être suspendu et une corticothérapie à la dose de 0,5 à 1 mg/kg/jour de méthylprednisolone ou équivalent doit être débutée. Après amélioration, ipilimumab en association à nivolumab peut être repris après réduction progressive des corticoïdes. En cas d'aggravation ou d'absence d'amélioration malgré l'initiation d'une corticothérapie, les doses de corticoïdes doivent être augmentées à 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent, et ipilimumab en association à nivolumab doit être arrêté définitivement.
Endocrinopathies d'origine-immunologique
Ipilimumab en association à nivolumab
Des endocrinopathies sévères, incluant hypothyroïdie, hyperthyroïdie, insuffisance surrénalienne (incluant l'insuffisance cortico-surrénalienne secondaire), hypophysite (incluant l'hypopituitarisme), diabète, et acidocétose diabétique ont été observées avec ipilimumab en association à nivolumab (voir rubrique Effets indésirables).
Les patients doivent être surveillés pour déceler l'apparition de signes et symptômes d'endocrinopathie, d'hyperglycémie et des modifications de la fonction thyroïdienne (au début du traitement, à intervalles réguliers en cours de traitement, et si cliniquement indiqué). Les patients peuvent présenter de la fatigue, des céphalées, des modifications de l'état mental, des douleurs abdominales, un transit intestinal inhabituel, et une hypotension, ou des symptômes non spécifiques qui peuvent faire penser à d'autres causes telles que des métastases cérébrales ou une maladie sous- jacente. A moins qu'une autre étiologie n'ait été identifiée, les signes et symptômes d'endocrinopathie doivent être considérés comme d'origine immunologique.
En cas d'hypothyroïdie symptomatique, ipilimumab en association à nivolumab doit être suspendu, et un traitement substitutif en hormone thyroïdienne doit être débuté, si nécessaire. En cas d'hyperthyroïdie symptomatique, ipilimumab en association à nivolumab doit être suspendu, et un traitement par antithyroïdiens doit être débuté, si nécessaire. Une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit également être envisagée, en cas de suspicion d'une inflammation aiguë de la thyroïde. Après amélioration, ipilimumab en association à nivolumab peut être repris si nécessaire après réduction progressive des corticoïdes. La surveillance de la fonction thyroïdienne doit être poursuivie afin de s'assurer que le traitement substitutif hormonal utilisé est approprié. Ipilimumab en association à nivolumab doit être arrêté définitivement en cas d'hyperthyroïdie ou d'hypothyroïdie pouvant menacer le pronostic vital.
En cas d'insuffisance surrénalienne symptomatique de Grade 2, ipilimumab en association à nivolumab doit être suspendu, et une corticothérapie substitutive à une dose physiologique doit être débutée, si nécessaire. Ipilimumab en association à nivolumab doit être arrêté définitivement en cas d'insuffisance surrénalienne sévère (Grade 3) ou pouvant menacer le pronostic vital (Grade 4). La surveillance de la fonction surrénalienne et des taux d'hormone doit être poursuivie afin de s'assurer que la corticothérapie substitutive appropriée est utilisée.
En cas d'hypophysite symptomatique de Grade 2 ou 3, ipilimumab en association à nivolumab doit être suspendu, et un traitement substitutif hormonal doit être débuté, si nécessaire. En cas de suspicion d'inflammation aiguë de la glande pituitaire, une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit également être envisagée. Après amélioration, ipilimumab en association à nivolumab peut être repris si nécessaire après réduction progressive des corticoïdes. Ipilimumab en association à nivolumab doit être arrêté définitivement en cas d'hypophysite menaçant le pronostic vital (Grade 4). La surveillance de la fonction pituitaire et des taux d'hormone doit être poursuivie afin de s'assurer que la corticothérapie substitutive utilisée est appropriée.
En cas de diabète symptomatique, ipilimumab en association à nivolumab doit être suspendu, et un traitement substitutif par insuline doit être débuté, si nécessaire. La surveillance de la glycémie doit être poursuivie afin d'assurer que le traitement substitutif par insuline utilisé est approprié. Ipilimumab en association à nivolumab doit être arrêté définitivement en cas de diabète menaçant le pronostic vital.
Réactions à la perfusion
Ipilimumab en association à nivolumab
Des réactions liées à la perfusion sévères ont été rapportées dans les essais cliniques d'ipilimumab en association à nivolumab (voir rubrique Effets indésirables). En cas de réaction à la perfusion sévère ou pouvant menacer le pronostic vital, la perfusion d'ipilimumab en association à nivolumab doit être arrêtée et un traitement médical approprié doit être administré. Les patients présentant une réaction à la perfusion d'intensité légère à modérée peuvent recevoir ipilimumab en association à nivolumab sous surveillance étroite et avec l'utilisation d'une prémédication suivant les recommandations locales de traitement pour la prophylaxie des réactions liées à la perfusion.
Autres effets indésirables d'origine-immunologique
Ipilimumab en association avec un inhibiteur du PD-1 ou PD-L1
On a observé une lymphohistiocytose hémophagocytaire (LHH) avec ipilimumab en association avec un inhibiteur PD-1 ou PD-L1 (y compris avec nivolumab) Des précautions doivent être prises lorsqu'ipilimumab est administré en association avec un inhibiteur PD-1 ou PD-L1. Si la LHH est confirmée, l'administration d'ipilimumab en association avec un inhibiteur PD-1 ou PD-L1 doit être interrompue et un traitement de la LHH doit être initié.
Ipilimumab en association à nivolumab
Les effets indésirables d'origine immunologique suivants ont été rapportés chez moins de 1% des patients traités par ipilimumab en association à nivolumab dans les essais cliniques quels que soient la dose et le type de tumeur : pancréatite, uvéite, démyélinisation, neuropathie autoimmune (incluant parésie des nerfs facial et abducens), syndrome de Guillain-Barré, myasthénie grave, syndrome myasthénique, méningite aseptique, encéphalite, gastrite, sarcoïdose, duodénite, myosite, myocardite et rhabdomyolyse. Des cas de syndrome de Vogt-Koyanagi-Harada et des décollements séreux de la rétine ont été rapportés après commercialisation (voir rubrique Effets indésirables). Une perte de vision transitoire a été rapportée chez des patients présentant une inflammation oculaire liée à ipilimumab.
En cas de suspicion d'effet indésirable d'origine immunologique, une évaluation appropriée doit être effectuée afin de confirmer l'étiologie ou d'exclure d'autres causes. En fonction de la gravité de l'effet indésirable, le traitement par ipilimumab en association à nivolumab doit être suspendu et des corticoïdes doivent être administrés. Après amélioration, ipilimumab en association à nivolumab peut être repris après réduction progressive des corticoïdes. Ipilimumab en association au nivolumab doit être définitivement arrêté en cas d'effet indésirable sévère récurrent d'origine immunologique, et pour tout effet indésirable d'origine immunologique mettant en jeu le pronostic vital.
Des cas de myotoxicité (myosite, myocardite et rhabdomyolyse), dont certains d'issue fatale, ont été rapportés avec ipilimumab en association à nivolumab. Si un patient développe des signes et symptômes de myotoxicité, une surveillance étroite doit être mise en place et le patient doit être adressé à un spécialiste pour évaluation et traitement sans délai. Sur la base de la sévérité de la myotoxicité, ipilimumab en association à nivolumab doit être suspendu ou arrêté (voir rubrique Posologie et mode d'administration), et un traitement approprié instauré.
Le diagnostic de myocardite exige un haut degré de suspicion. Les patients présentant des symptômes cardiaques ou cardio-pulmonaires doivent être évalués pour une myocardite potentielle. Si une myocardite est suspectée, l'administration immédiate d'une dose élevée de stéroïdes (prednisone 1 à 2 mg/kg/jour ou méthylprednisolone 1 à 2 mg/kg/jour) et une consultation immédiate en cardiologie avec un bilan diagnostique selon les directives cliniques actuelles, doivent être initiées. Une fois qu'un diagnostic de myocardite est établi, le traitement par ipilimumab en association à nivolumab doit être suspendu ou définitivement interrompu (voir rubrique Posologie et mode d'administration).
Précautions spécifiques à la maladie
Mésothéliome pleural malin
Les patients présentant un mésothéliome primitif péritonéal, péricardique, ou de la tunique vaginale des testicules, une maladie pulmonaire interstitielle, une maladie auto-immune active, toute maladie nécessitant une immunosuppression systémique, et des métastases cérébrales (à moins d'une résection chirurgicale ou d'une radiothérapie stéréotaxique, et sans évolution dans les 3 mois précédant l'inclusion dans l'étude) ont été exclus de l'étude pivot dans le traitement de première ligne du MPM (voir rubriques Interactions avec d'autres médicaments et autres formes d'interactions et Propriétés pharmacodynamiques). En l'absence de données, l'ipilimumab en association au nivolumab doit être utilisé avec précaution dans ces populations, après évaluation attentive du bénéfice/risque potentiel au cas par cas.
Patients atteints de maladie auto-immune
Les patients ayant des antécédents de maladie auto-immune (mis à part les patients atteints de vitiligo et de déficiences endocriniennes correctement contrôlées tel qu'une hypothyroïdie), y compris ceux devant recevoir un traitement immunosuppresseur par voie systémique pour traiter une maladie auto- immune préexistante ou suite à une greffe d'organe n'ont pas été étudiés lors des essais cliniques.
Ipilimumab est un potentialisateur des cellules-T qui stimule la réponse immunitaire (voir
rubrique Propriétés pharmacodynamiques) et peut interférer avec la thérapie immunosuppressive, entrainant une exacerbation de la pathologie sous-jacente ou une augmentation du risque de rejet de greffe. Ipilimumab doit être évité chez les patients souffrant d'une maladie auto-immune active sévère, pour laquelle une activation immunologique supplémentaire pourrait potentiellement et de façon imminente menacer le pronostic vital. Chez les autres patients ayant des antécédents de maladie auto-immune, ipilimumab doit être utilisé avec précaution après avoir minutieusement évalué le rapport bénéfice-risque individuel potentiel.
Patients sous régime hyposodé contrôlé
Ce médicament contient 23 mg de sodium par flacon de 10 ml, équivalent à 1,15% de l'apport quotidien maximal recommandé par l'OMS de 2 g de sodium pour un adulte. A prendre en compte lors du traitement des patients suivant un régime hyposodé contrôlé.
Ipilimumab en association à nivolumab (voir section Posologie et mode d'administration)
a. Résumé du profil de sécurité
Lorsqu'ipilimumab est administré en association à nivolumab, se référer au Résumé des Caractéristiques du Produit de nivolumab avant l'initiation du traitement. Pour des informations complémentaires sur les mises en garde et précautions d'emploi associé au traitement nivolumab, se référer au RCP de nivolumab.
MPM
Dans l'ensemble des données d'ipilimumab 1 mg/kg en association à nivolumab 3 mg/kg dans le MPM (n = 300), avec un suivi minimum de 17,4 mois, les effets indésirables les plus fréquents (≥ 10%) ont été : fatigue (43%), diarrhée (31%), éruption cutanée (30%), douleur musculosquelettique (27%), nausée (24%), diminution de l'appétit (24%), prurit (21%), constipation (19%), et hypothyroïdie (13%).
La plupart des effets indésirables était d'intensité légère à modérée (Grade 1 ou 2).Liste tabulée des effets indésirables
Les effets indésirables rapportés dans l'ensemble des données poolées chez les patients traités par l'ipilimumab 1 mg/kg en association à nivolumab 3 mg/kg dans le MPM (n = 300) sont présentés dans le Tableau 2. Ces effets sont présentés par classe de systèmes d'organes et par ordre de fréquence. Les fréquences sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données post-commercialisation disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre de gravité décroissant.
| Ipilimumab 1 mg/kg en association à Nivolumab 3 mg/kg dans le MPM*** | |
| Affections du système immunitaire | |
| Fréquent | réaction liée à la perfusion, hypersensibilité |
| Affections endocriniennes | |
| Très fréquent | hypothyroïdie |
| Fréquent | hyperthyroïdie, insuffisance surrénalienne, hypophysite, hypopituitarisme |
| Peu fréquent | thyroïdite |
| Troubles du métabolisme et de la nutrition | |
| Très fréquent | diminution de l'appétit |
| Affections du système nerveux | |
| Peu fréquent | encéphalite |
| Affections cardiaques | |
| Peu fréquent | myocardite |
| Affections respiratoires, thoraciques et médiastinales | |
| Fréquent | pneumopathie inflammatoire |
| Affections gastro-intestinales | |
| Très fréquent | diarrhée, nausée, constipation |
| Fréquent | colite, pancréatite |
| Affections hépatobiliaires | |
| Fréquent | hépatite |
| Affections de la peau et du tissu sous-cutané | |
| Très fréquent | éruption cutanéeb, prurit |
| Affections musculo-squelettiques et systémiques | |
| Très fréquent | douleur musculo-squelettiquec |
| Fréquent | arthrite |
| Peu fréquent | myosite |
| Affections du rein et des voies urinaires | |
| Fréquent | atteinte rénale aiguë, insuffisance rénale |
| Troubles généraux et anomalies au site d'administration | |
| Très fréquent | fatigue |
| Investigationsb | |
| Très fréquent | augmentation du taux d'ASAT, augmentation du taux d'ALAT, augmentation du taux de phosphatases alcalines, augmentation de la lipase, augmentation de l'amylase, augmentation du taux de créatinine, hyperglycémiea, lymphopénie, anémie, hypercalcémie, hypocalcémie, hyperkaliémie, hypokaliémie, hyponatrémie, hypomagnésémie |
| Fréquent | augmentation du taux de bilirubine totale, hypoglycémie, leucopénie, neutropéniea, thrombocytopénie, hypernatrémie, hypermagnésémie |
*** ipilimumab en association à nivolumab dans le MPM.
a Des cas d'issue fatale ont été rapportés dans les études cliniques terminées ou en cours.
b Eruption cutanée est un terme composite incluant éruption cutanée macropapuleuse, éruption cutanée érythémateuse, éruption cutanée prurigineuse, éruption cutanée folliculaire, éruption cutanée maculaire, éruption cutanée morbiliforme, éruption cutanée papuleuse, éruption cutanée pustuleuse, éruption cutanée papulosquameuse, éruption cutanée vésiculaire, éruption cutanée généralisée, éruption cutanée exfoliative, dermatite, dermatite acnéiforme, dermatite allergique, dermatite atopique, dermatite bulleuse, dermatite exfoliative, dermatite psoriasiforme, éruption d'origine médicamenteuse et pemphigoïde.
c Douleur musculosquelettique est un terme composite qui inclut douleur dorsale, douleur osseuse, douleur de type musculosquelettique dans le thorax, gêne musculosquelettique, myalgie, douleur cervicale, douleur des extrémités et douleur rachidienne.
Description de certains effets indésirables sélectionnés
Ipilimumab en association au nivolumab est associé à des effets indésirables d'origine immunologique. Avec un traitement médical approprié, les effets indésirables d'origine immunologique ont été résolus dans la plupart des cas. L'arrêt définitif du traitement a été nécessaire chez une plus grande proportion de patients recevant ipilimumab en association à nivolumab que chez ceux recevant nivolumab en monothérapie. Le Tableau 3 présente le pourcentage de patients ayant eu des effets indésirables d'origine immunologique et qui ont définitivement arrêté le traitement en fonction du schéma posologique. De plus, pour les patients ayant eu un évènement, le Tableau 3 présente le pourcentage de patients ayant nécessité des corticoïdes à haute dose (au moins 40 mg d'équivalent prednisone par jour) en fonction du schéma posologique. Les recommandations de prise en charge de ces effets indésirables sont décrites au niveau de la rubrique Mises en garde et précautions d'emploi.
- Tableau 3: Effets indésirables d'origine immunologique entraînant l'arrêt définitif ou nécessitant des doses élevées de corticoïdes, en fonction du schéma posologique
| | Ipilimumab 1 mg/kg en association à nivolumab 3 mg/kg dans le MPM % |
| Effet indésirable d'origine immunologique entraînant l'arrêt définitif | |
| Pneumopathie inflammatoire | 2,3 |
| Colite | 5,0 |
| Hépatite | 3,7 |
| Néphrite et dysfonctionnement rénal | 1,3 |
| Endocrinopathies | 0,3 |
| Peau | 0,7 |
| Hypersensibilité/Réaction à la perfusion | 1,7 |
| Effet indésirable d'origine immunologique nécessitant des doses élevées de corticoïdesa,b | |
| Pneumopathie inflammatoire | 70 |
| Colite | 33 |
| Hépatite | 42 |
| Néphrite et dysfonctionnement rénal | 40 |
| Endocrinopathies | 10 |
| Peau | 8 |
| Hypersensibilité/Réaction à la perfusion | 17 |
a Au moins 40 mg d'équivalent prednisone par jour
b La fréquence est basée sur le nombre de patients qui ont présenté l'effet indésirable d'origine immunologique.
Colite d'origine immunologique
Chez les patients traités par l'ipilimumab 1 mg/kg en association à nivolumab 3 mg/kg dans le MPM, l'incidence des diarrhées ou colites était de 22,0% (66/300). Des cas de Grade 2 et Grade 3 ont été rapportés chez respectivement 7,3% (22/300) et 5,3% (16/300) des patients. Le délai médian de survenue était de 3,9 mois (de 0,0 à 21,7 mois). Une résolution est survenue chez 62 patients (93,9%) avec un délai médian de résolution de 3,1 semaines (de 0,1 à 100,0+ semaines).
Pneumopathie inflammatoire d'origine immunologique
Chez les patients traités par l'ipilimumab 1 mg/kg en association à nivolumab 3 mg/kg dans le MPM, l'incidence des pneumopathies inflammatoires, incluant des pneumopathies interstitielles, était de 6,7% (20/300). Des cas de Grade 2 et Grade 3 ont été rapportés chez respectivement 5,3% (16/300) et 0,7% (2/300) des patients. Le délai médian de survenue était de 1,8 mois (de 0,3 à 20,8 mois).
Une résolution est survenue chez 16 patients (80%) avec un délai médian de résolution de 6,1 semaines (de 1,1 à 113,1+ semaines).
Hépatite d'origine immunologique
Chez les patients traités par l'ipilimumab 1 mg/kg en association à nivolumab 3 mg/kg dans le MPM, l'incidence des anomalies de la fonction hépatique était de 12,0% (36/300). Des cas de Grade 2, Grade 3 et Grade 4 ont été rapportés chez respectivement 1,7% (5/300), 4,3% (13/300) et 1,0% (3/300) des patients. Le délai médian de survenue était de 1,8 mois (de 0,5 à 20,3 mois). Une résolution est survenue chez 31 patients (86,1%) avec un délai médian de résolution de 4,1 semaines (de 1,0 à 78,3+ semaines).
Effets indésirables cutanés d'origine immunologique
Chez les patients traités par l'ipilimumab 1 mg/kg en association à nivolumab 3 mg/kg dans le MPM, l'incidence des éruptions cutanées était de 36,0% (108/300). Des cas de Grade 2 et Grade 3 ont été rapportés chez respectivement 10,3% (31/300) et 3,0% (9/300) des patients. Le délai médian de survenue était de 1,6 mois (de 0,0 à 22,3 mois). Une résolution est survenue chez 71 patients (66,4%) avec un délai médian de résolution de 12,1 semaines (de 0,4 à 146,4+ semaines).
Néphrite et dysfonction rénale d'origine immunologique
Chez les patients traités par l'ipilimumab 1 mg/kg en association à nivolumab 3 mg/kg dans le MPM, l'incidence des dysfonctions rénales était de 5,0% (15/300). Des cas de Grade 2 et de Grade 3 ont été rapportés chez respectivement 2,0% (6/300) et 1,3% (4/300) des patients. Le délai médian de survenue était de 3,6 mois (de 0,5 à 14,4). Une résolution est survenue chez 12 patients (80,0%) avec un délai médian de résolution de 6,1 semaines (de 0,9 à 126,4+).
Endocrinopathies d'origine immunologique
Chez les patients traités par l'ipilimumab 1 mg/kg en association à nivolumab 3 mg/kg dans le MPM, l'incidence des troubles thyroïdiens était de 14% (43/300). Des troubles thyroïdiens de Grade 2 et de Grade 3 ont été rapportés chez respectivement 9,3% (28/300) et 1,3% (4/300) des patients. Des hypophysites sont survenues chez 2% (6/300) des patients. Des cas de Grade 2 ont été rapportés chez 1,3% (4/300) des patients. Des hypopituitarismes de Grade 2 et de Grade 3 sont survenus chez respectivement 1,0% (3/300) et 1,0% (3/300) des patients. Des insuffisances surrénaliennes de Grade 2 et Grade 3 sont survenues chez respectivement 1,7% (5/300) et 0,3% (1/300) des patients. Le délai médian de survenue de ces endocrinopathies était de 2,8 mois (de 0,5 à 20,8 mois). Une résolution est survenue chez 17 patients (32,7%). Le délai de résolution allait de 0,3 à 144,1+ semaines.
Réactions à la perfusion
Chez les patients traités par l'ipilimumab 1 mg/kg en association à nivolumab 3 mg/kg dans le MPM, l'incidence des hypersensibilités/réactions à la perfusion était de 12% (36/300). Des cas de Grade 2 et de Grade 3 ont été rapportés chez respectivement 5,0% (15/300) et 1,3% (4/300) des patients.
Immunogénicité
Chez les patients traités par ipilimumab en association à nivolumab et évaluables pour la présence d'anticorps anti-ipilimumab, l'incidence des anticorps anti-ipilimumab allait de 6,3 à 13,7%. Les anticorps neutralisants dirigés contre ipilimumab allaient de 0 à 0,4%. Chez les patients évaluables pour la présence d'anticorps anti-nivolumab, l'incidence des anticorps anti-nivolumab était de 26% avec nivolumab 3mg/kg et ipilimumab 1 mg/kg toutes les 3 semaines, 25,7% avec nivolumab 3mg/kg toutes les 2 semaines et ipilimumab 1 mg/kg toutes les 6 semaines, et 37,8% avec nivolumab 1 mg/kg et ipilimumab 3 mg/kg toutes les 3 semaines. L'incidence des anticorps neutralisants dirigés contre nivolumab était de 0,5% avec nivolumab 3mg/kg et ipilimumab 1 mg/kg toutes les 3 semaines, 0,7% avec nivolumab 3mg/kg toutes les 2 semaines et ipilimumab 1 mg/kg toutes les 6 semaines, et 4,6% avec nivolumab 1 mg/kg et ipilimumab 3mg/kg toutes les 3 semaines.
Lorsqu'administré en association à nivolumab, la clairance d'ipilimumab était inchangée en présence des anticorps anti-ipilimumab et il n'avait pas été mis en évidence d'altération du profil de tolérance.
Anomalies des valeurs biologiques
Chez les patients traités par l'ipilimumab 1 mg/kg en association à nivolumab 3 mg/kg dans le MPM, la proportion de patients ayant présenté une aggravation des paramètres biologiques par rapportà l'inclusion vers une anomalie de Grade 3 ou 4 a été la suivante : 2,4% pour les anémies, 1,0% chacune pour les thrombocytopénies et les leucopénies, 8,4% pour les lymphopénies, 1,3% pour les neutropénies, 3,1% pour les augmentations du taux de phosphatases alcalines, 7,1% chacune pour les augmentations du taux d'ASAT et du taux d'ALAT, 1,7% pour les augmentations du taux de bilirubine totale, 0,3% pour les augmentations du taux de créatinine, 2,8% pour les hyperglycémies, 5,4% pour les augmentations de l'amylase, 12,8% pour l'augmentation de la lipase, 0,7% pour les hypernatrémies, 8,1% pour les hyponatrémies, 4,1% pour les hyperkaliémies, 2,0% pour les hypokaliémies et 0,3% pour les hypocalcémies.
Patients âgés
Dans le MPM, il y a eu un taux plus important d'effets indésirables graves et d'arrêts de traitement dus à un effet indésirable chez les patients âgés de 75 ans ou plus (68% et 35% respectivement) comparé à tous les patients ayant reçu du nivolumab en association à l'ipilimumab (54% et 28% respectivement).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : signalement.social-sante.gouv.fr.
SURVEILLANCE du traitement :
- Fonctions hépatique et thyroïdienne à évaluer avant l'instauration du traitement et avant chaque administration.
- Surveillance continue des effets indésirables cardiaques et
pulmonaires, ainsi que pour des signes cliniques, des symptômes et des
anomalies biologiques indiquant des troubles électrolytiques et une
déshydratation, avant l'initiation du traitement et à intervalles
réguliers au cours du traitement
Grossesse
Il n'existe pas de données sur l'utilisation de YERVOY chez la femme enceinte. Les études de reproduction effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique Données de sécurité précliniques). L'IgG1 humaine traverse la barrière placentaire. Le risque potentiel du traitement pour le foetus en cours de développement n'est pas connu. YERVOY ne doit pas être utilisé pendant la grossesse ou chez les femmes susceptibles de procréer n'utilisant pas une méthode efficace de contraception, à moins que le bénéfice clinique attendu soit supérieur au risque potentiel.
Allaitement
De très faibles concentrations d'ipilimumab ont été retrouvées dans le lait maternel chez des singes Cynomolgus traités pendant la grossesse. On ignore si ipilimumab est excrété dans le lait humain. La sécrétion des l'IgGs dans le lait humain est généralement limitée et les IgGs ont une faible biodisponibilité par voie orale. Une exposition systémique importante du nourrisson n'est pas attendue et aucun effet de l'allaitement sur le nouveau-né/nourrisson n'est anticipé. Cependant, étant donné le risque potentiel d'effet indésirable pour l'enfant allaité, il faudra décider soit d'arrêter l'allaitement soit d'arrêter le traitement par YERVOY, en tenant compte du bénéfice de l'allaitement pour l'enfant et du bénéfice du traitement pour la mère.
Fertilité
Aucune étude n'a été menée pour évaluer l'effet d'ipilimumab sur la fertilité. Par conséquent, l'effet d'ipilimumab sur la fertilité masculine ou féminine n'est pas connu.
Ipilimumab est un anticorps monoclonal humain qui n'est pas métabolisé par les enzymes du cytochrome P450 (CYPs) ou d'autres enzymes métabolisant les médicaments.
Une étude d'interaction médicamenteuse menée chez les adultes avec ipilimumab administré seul et en association avec une chimiothérapie (dacarbazine ou paclitaxel/carboplatine) a été conduite pour évaluer l'interaction avec les isoenzymes CYP (particulièrement CYP1A2, CYP2E1, CYP2C8 et CYP3A4) chez des patients atteints d'un mélanome avancé naïfs de traitement. Aucune interaction pharmacocinétique médicamenteuse cliniquement pertinente n'a été observée entre ipilimumab et paclitaxel/carboplatine, dacarbazine ou son métabolite, le 5-aminoimidazole-4-carboxamide (AIC).
Autres formes d'interaction
Corticostéroïdes
Avant l'instauration d'un traitement par ipilimumab, l'utilisation de corticostéroïdes systémiques doit être évitée car ils pourraient interférer avec l'activité pharmacodynamique et l'efficacité d'ipilimumab.
Néanmoins, les corticostéroïdes systémiques ou d'autres immunosuppresseurs peuvent être utilisés après l'instauration d'un traitement par ipilimumab pour traiter les effets indésirables d'origine-immunologique. L'utilisation de corticostéroïdes systémiques après l'instauration d'un traitement par ipilimumab ne paraît pas altérer l'efficacité d'ipilimumab.
Anticoagulants
L'utilisation d'anticoagulants est connue pour augmenter le risque d'hémorragie gastrointestinale. L'augmentation du risque d'hémorragie gastrointestinale par l'utilisation d'anticoagulants est établie. Etant donné que l'hémorragie gastrointestinale est un effet indésirable d'ipilimumab (voir section 4.8), les patients nécessitant un traitement anticoagulant concomitant doivent être surveillés étroitement.
Le traitement doit être initié et surveillé par des médecins spécialistes expérimentés dans le traitement du cancer.
Posologie
YERVOY en association à nivolumab
Mésothéliome Pleural Malin
La dose recommandée est de 1 mg/kg d'ipilimumab administrée par voie intraveineuse pendant 30 minutes toutes les 6 semaines, en association avec 360 mg de nivolumab administrée par voie intraveineuse pendant 30 minutes toutes les 3 semaines. Le traitement est poursuivi jusqu'à 24 mois chez les patients sans progression de la maladie.
Durée du traitement
Le traitement par YERVOY en association au nivolumab doit être poursuivi tant qu'un bénéfice clinique est observé ou jusqu'à ce que le patient ne puisse plus tolérer le traitement (et jusqu'à la durée maximale du traitement si celle-ci est spécifiée pour une indication).
Des réponses atypiques (c'est à dire une augmentation initiale transitoire de la taille de la tumeur ou l'apparition de nouvelles petites lésions au cours des premiers mois, suivi de réduction de la tumeur) ont été observées. Il est recommandé de continuer le traitement par YERVOY en association au nivolumab chez les patients cliniquement stables présentant des signes initiaux de progression de la maladie jusqu'à ce que la progression de la maladie soit confirmée.
Les fonctions hépatique et thyroïdienne doivent être évaluées avant l'instauration d'un traitement par YERVOY et avant chaque administration. De plus, tout signe ou symptôme évocateur d'effets indésirables immunologiques, tels que diarrhée et colite, doit être évalué pendant le traitement par YERVOY (voir la rubrique Mises en garde et précautions d'emploi).
Arrêt définitif du traitement ou suspension de doses
La prise en charge d'effets indésirables immunologiques peut nécessiter la suspension ou l'arrêt définitif du traitement par YERVOY et l'instauration d'une corticothérapie systémique à hautes doses. Dans certains cas, l'ajout d'un traitement immunosuppresseur est à envisager (voir rubrique Mises en garde et précautions d'emploi).
Les augmentations ou les diminutions de doses ne sont pas recommandées. Des administrations différées ou des interruptions de traitement peuvent être nécessaires selon la tolérance individuelle et l'innocuité du traitement.
Les recommandations d'arrêt définitif du traitement ou de suspension de doses sont décrites dans le Tableau 1 pour YERVOY en association au nivolumab. Les recommandations détaillées de la prise en charge des effets indésirables d'origine immunologique sont décrites à la rubrique Mises en garde et précautions d'emploi.
Tableau 1: Recommandations de modification du traitement par YERVOY en association à nivolumab ou pour l'administration de la seconde phase de traitement (nivolumab en monothérapie) suivant la phase d'association
![]()
![]() Effets indésirables d'origine immunologique
Effets indésirables d'origine immunologique
![]() Pneumopathie inflammatoire d'origine immunologique
Pneumopathie inflammatoire d'origine immunologique
Colite d'origine immunologique
Sévérité Modification de traitement
Pneumopathie de Grade 2 Suspendre la(les) dose(s) jusqu'à la
résolution des symptômes, l'amélioration des anomalies radiographiques, et la fin du traitement par corticoïdes
Pneumopathie de Grade 3 ou 4 Arrêt définitif du traitement
Diarrhée ou colite de Grade 2 Suspendre la(les) dose(s) jusqu'à la
résolution des symptômes et la fin du traitement par corticoïdes, s'il s'est avéré nécessaire
Diarrhée ou colite de grade 3 ou 4 Arrêt définitif du traitement
![]()
Hépatite d'origine immunologique
Néphrite et dysfonction rénale d'origine immunologique
Endocrinopathies d'origine immunologique
Elévation de Grade 2 des aspartate aminotransférases (ASAT), des alanine aminotransférases (ALAT), ou de la bilirubine totale
![]() Elévation de Grade 3 ou 4 des ASAT, ALAT, ou de la bilirubine totale
Elévation de Grade 3 ou 4 des ASAT, ALAT, ou de la bilirubine totale
Elévation de la créatininémie de Grade 2 ou 3
![]() Elévation de la créatininémie de Grade 4
Elévation de la créatininémie de Grade 4
Hypothyroïdie, hyperthyroïdie, hypophysite de Grade 2 ou 3 symptomatiques,
Insuffisance surrénalienne de Grade 2 Diabète de Grade3
Suspendre la(les) dose(s) jusqu'au retour des valeurs biologiques aux valeurs initiales et jusqu'à la fin du traitement par corticoïdes, s'il s'est avéré nécessaire
Arrêt définitif du traitement
Suspendre la(les) dose(s) jusqu'au retour de la créatininémie à la valeur initiale et jusqu'à la fin du traitement par corticoïdes
Arrêt définitif du traitement
Suspendre la(les) dose(s) jusqu'à la résolution des symptômes et la fin du traitement par corticoïdes (s'il s'est avéré nécessaire pour les symptômes d'une inflammation aiguë). Le traitement doit être
Hypothyroïdie de Grade 4 Hyperthyroïdie de Grade 4 Hypophysite de Grade 4
Insuffisance surrénalienne de Grade 3 ou 4
![]() Diabète de Grade 4
Diabète de Grade 4
maintenu en cas de traitement substitutif hormonala tant qu'il n'y a pas de présence de symptômes
![]() Arrêt définitif du traitement
Arrêt définitif du traitement
Effets indésirables
cutanés d'origine immunologique
Eruption cutanée de Grade 3 Suspendre la(les) dose(s) jusqu'à la
résolution des symptômes et la fin du traitement par corticoïdes
Eruption cutanée de Grade 4 Arrêt définitif du traitement
Syndrome de Stevens-Johnson (SJS) ou nécrolyse épidermique toxique (NET)
Arrêt définitif du traitement (voir rubrique Mises en garde et précautions d'emploi)
Myocardite d'origine immunologique
Myocardite de Grade 2 Suspendre la(les) dose(s) jusqu'à la résolution des symptômes et la fin du traitement par corticoïdes b
![]() Myocardite
de Grade 3 ou
4
Arrêt définitif du traitement Grade 3 (première
apparition)
Suspendre la(les) dose(s)
Myocardite
de Grade 3 ou
4
Arrêt définitif du traitement Grade 3 (première
apparition)
Suspendre la(les) dose(s)
Autres effets
indésirables d'origine immunologique
Grade 4 ou Grade 3 récidivant ; Grade 2 ou 3 persistant malgré une modification de traitement ; impossibilité de réduire la dose de corticoïdes à 10 mg de prednisone ou équivalent par jour
Arrêt définitif du traitement
![]()
Note: Les grades de toxicité correspondent à la classification du National Cancer Institute Common terminology Criteria for Adverse Events Version 4.0 (NCI-CTCAE v4).
a La recommandation pour l'utilisation d'un traitement substitutif hormonal est fournie en rubrique Mises en garde et précautions d'emploi.
b La tolérance de la reprise du traitement par ipilimumab en association à nivolumab, chez les patients ayant présentés précédemment une myocardite d'origine immunologique, n'est pas connue.
YERVOY en association à nivolumab doit être définitivement arrêté en cas de :
· Effets indésirables de Grade 4 ou de Grade 3 récidivants;
· Effets indésirables de Grade 2 ou3 persistants malgré leur prise en charge.
Lorsque YERVOY est administré en association à nivolumab, si l'un des traitements est suspendu, l'autre traitement devra aussi être suspendu. Si l'administration est reprise après un temps différé, soit le traitement en association ou nivolumab en monothérapie peut être repris sur la base de l'évaluation individuelle du patient.
Populations particulières
Population pédiatrique
La sécurité et l'efficacité de YERVOY chez les enfants de moins de 12 ans n'ont pas été établies. Les données disponibles sont très limitées. YERVOY ne doit pas être utilisé chez les enfants de moins de 12 ans.
Personnes âgées
Aucune différence en termes de sécurité ou d'efficacité n'a été rapportée entre les patients âgé (≥ 65 ans) et les patients plus jeunes (< 65 ans) traités par YERVOY. Aucune adaptation posologique n'est nécessaire dans cette population (voir rubrique Propriétés pharmacodynamiques).
Insuffisance rénale
La sécurité et l'efficacité de YERVOY chez les insuffisants rénaux n'ont pas été étudiées. D'après les résultats pharmacocinétiques dans la population étudiée, aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance rénale légère à modérée (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
La sécurité et l'efficacité de YERVOY chez les patients présentant une insuffisance hépatique n'ont pas été étudiées. Sur la base des données de pharmacocinétique de population, aucun ajustement de dose spécifique n'est nécessaire chez les patients présentant une insuffisance hépatique légère (voir rubrique Propriétés pharmacocinétiques). YERVOY doit être administré avec prudence chez les patients dont les taux de transaminases sont ≥ 5 x LSN ou les taux de bilirubine sont > 3 x LSN à l'état de base (voir rubrique Propriétés pharmacodynamiques).
Mode d'administration
YERVOY est pour usage intraveineux. Le temps de perfusion recommandé est de 30 minutes.
YERVOY peut être utilisé en administration intraveineuse sans dilution ou être dilué dans une solution injectable de chlorure de sodium à 9 mg/ml (0,9%) ou de glucose à 50 mg/ml (5%) à des concentrations de 1 mg/ml à 4 mg/ml et conservé dans des flacons en verre ou des poches en PVC ou sans PVC.
YERVOY ne doit pas être injecté en IV directe rapide ni en bolus IV.
Lorsqu'il est administré en association à nivolumab, nivolumab doit être administré en premier suivi par YERVOY le même jour. Utiliser des poches et des filtres de perfusion distincts pour chaque perfusion.
Pour les instructions concernant la préparation et la manipulation du médicament avant administration, voir la rubrique Instructions pour l'utilisation, la manipulation et l'élimination.
Durée de conservation :
Flacon non ouvert 3 ans
Après ouverture
D'un point de vue microbiologique, une fois ouvert, le produit doit être perfusé ou dilué puis perfusé immédiatement. La stabilité chimique et physique en cours d'utilisation de la solution diluée ou non (entre 1 et 4 mg/ml) a été démontrée pendant 24 heures à 25°C et entre 2°C et 8°C. Si elle n'est pas utilisée immédiatement, la solution pour perfusion (diluée ou non) peut être conservée au réfrigérateur jusqu'à 24 heures (entre 2°C et 8°C) ou à température ambiante (entre 20°C et 25°C).
Précautions particulières de conservation :
A conserver au réfrigérateur (entre 2°C et 8°C). Ne pas congeler.
A conserver dans l'emballage extérieur d'origine à l'abri de la lumière.
Pour les conditions de conservation du médicament après première ouverture ou dilution, voir la rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
La dose maximale tolérée d'ipilimumab n'a pas été déterminée. Dans les essais cliniques, les patients ont reçu jusqu'à 20 mg/kg sans effet toxique apparent.
En cas de surdosage, les patients doivent être étroitement surveillés à la recherche de signes ou symptômes évocateurs d'effets indésirables, et un traitement symptomatique approprié doit être instauré.
Classe pharmacothérapeutique : Agents antinéoplasiques, anticorps monoclonaux code ATC : L01XC11.
Mécanisme d'action
L'antigène 4 des lymphocytes T cytotoxiques (CTLA-4) est un régulateur majeur de l'activité des cellules T. Ipilimumab est un inhibiteur du point de contrôle immunitaire CTLA-4 qui bloque les signaux inhibiteurs des cellules T induits par la voie du CTLA-4, en augmentant le nombre de cellules T-effectrices réactives qui se mobilisent pour augmenter l'attaque immunologique directe des cellules T contre les cellules tumorales. Le blocage du CTLA-4 peut également réduire la fonction des cellules T régulatrices, ce qui peut contribuer à une réponse immunitaire anti-tumorale. Ipilimumab peut réduire sélectivement les cellules T régulatrices au niveau du site de la tumeur, conduisant à une augmentation du ratio intratumoral cellules T effectrices / cellules T régulatrices, qui entraîne la mort des cellules tumorales.
Effets pharmacodynamiques
Chez les patients atteints de mélanome ayant reçu ipilimumab, le taux moyen de lymphocytes dans le sang circulant a augmenté au cours de la période d'induction. Au cours des études de Phase 2, cette augmentation était dose-dépendante. Au cours de l'étude MDX010-20 (voir rubrique Propriétés pharmacodynamiques), l'administration d'ipilimumab à la dose de 3 mg/kg associé ou non au gp100 a permis une augmentation du taux de lymphocytes dans le sang circulant au cours de la période d'induction, tandis qu'aucun changement significatif de ce taux n'a été observé dans le groupe de patients contrôle ne recevant que le vaccin peptidique expérimental gp100 seul.
Dans le sang périphérique des patients atteints de mélanome, une augmentation moyenne du pourcentage de cellules T activées HLA-DR+ CD4+ et CD8+ a été observée après traitement par ipilimumab, ce qui est en accord avec son mécanisme d'action. Une hausse moyenne du pourcentage de cellules T à mémoire centrale (CCR7+ CD45RA-) CD4+ et CD8+ T et une augmentation moyenne plus faible, mais significative, du pourcentage de cellules T à mémoire effectrice (CCR7- CD45RA-) CD8+ ont également été observées après traitement par ipilimumab.
Efficacité et sécurité clinique
YERVOY en association à nivolumab
Pour plus d'informations sur l'efficacité et la sécurité clinique associées aux recommandations posologiques de nivolumab en monothérapie administré à la suite d'un traitement en association à l'ipilimumab, se référer au RCP de nivolumab.
Sur la base d'un modèle de relations dose/réponse de l'efficacité et de la tolérance, il n'y a pas de différences cliniquement significatives en termes d'efficacité et de tolérance entre la posologie de nivolumab à 240 mg toutes les 2 semaines et à 3 mg/kg toutes les 2 semaines. De plus, sur la base de ces relations, il n'y a pas eu de différences cliniquement significatives entre la posologie de nivolumab à 480 mg toutes les 4 semaines et à 3 mg/kg toutes les 2 semaines dans le mélanome avancé et le CCR.
Essais cliniques avec ipilimumab en association avec nivolumab
Mésothéliome pleural malin
Étude randomisée de phase 3 avec nivolumab en association à l'ipilimumab versus chimiothérapie (CA209743)
La tolérance et l'efficacité de l'ipilimumab 1 mg/kg toutes les 6 semaines en association à nivolumab 3 mg/kg toutes les 2 semaines ont été évaluées dans une étude randomisée de phase 3, en ouvert (CA209743). L'étude a inclus des patients (âgés de 18 ans ou plus) avec un mésothéliome pleural malin histologiquement confirmé et non préalablement traité, d'histologie épithélioïde ou non- épithélioïde, avec un statut de performance ECOG de 0 ou 1, et sans radiothérapie palliative dans les 14 jours précédant le premier traitement à l'étude. Les patients ont été inclus indépendamment du statut PD-L1 de leur tumeur.
Les patients présentant un mésothéliome primitif péritonéal, péricardique, ou de la tunique vaginale des testicules, une maladie pulmonaire interstitielle, une maladie auto-immune active, toute maladie nécessitant une immunosuppression systémique, et des métastases cérébrales (à moins d'une résection chirurgicale ou d'une radiothérapie stéréotaxique, et sans évolution dans les 3 mois précédant l'inclusion dans l'étude) ont été exclus de l'étude. La randomisation a été stratifiée en fonction de l'histologie (sous-types épithélioïdes vs. sarcomatoïdes ou histologie mixte) et du sexe (masculin vs. féminin).
Un total de 605 patients a été randomisé pour recevoir de l'ipilimumab en association au nivolumab (n = 303) ou une chimiothérapie (n = 302). Les patients du bras ipilimumab en association au nivolumab ont reçu de l'ipilimumab 1 mg/kg, administré par voie intraveineuse pendant 30 minutes toutes les 6 semaines en association au nivolumab 3 mg/kg, administré par voie intraveineuse pendant 30 minutes toutes les 2 semaines pour une durée maximale de 2 ans. Les patients du bras chimiothérapie ont reçu une chimiothérapie pendant un maximum de 6 cycles (chaque cycle était de 21 jours). La chimiothérapie comprenait du cisplatine 75 mg/m² et du pemetrexed 500 mg/m², ou du carboplatine (ASC de 5) et du pemetrexed 500 mg/m².
Le traitement a été poursuivi jusqu'à progression de la maladie, toxicité inacceptable ou jusqu'à 24 mois. Le traitement pouvait se poursuivre après progression de la maladie si le patient était cliniquement stable et que l'investigateur considérait qu'il en tirait un bénéfice clinique. Les patients qui ont arrêté le traitement en association en raison d'un événement indésirable attribué à l'ipilimumab ont étaient autorisés à poursuivre le nivolumab en monothérapie. Les évaluations tumorales ont été réalisées toutes les 6 semaines après la première dose du traitement à l'étude pendant les 12 premiers mois, puis toutes les 12 semaines et ce jusqu'à progression de la maladie ou arrêt du traitement à l'étude.
Les caractéristiques des patients à l'inclusion dans l'étude CA209743 étaient généralement équilibrées entre les différents bras de traitement. L'âge médian était de 69 ans (de 25 à 89 ans), avec 72% ≥ 65 ans et 26% ≥ 75 ans. La majorité des patients était de type caucasien (85%) et de sexe masculin (77%). Le statut de performance ECOG à l'inclusion était de 0 (40%) ou de 1 (60%), 80% des patients présentaient un statut PD-L1 ≥ 1% et 20% un statut PD-L1 < 1%, 75% présentaient une histologie épithélioïde et 25% non épithélioïde.
Le critère d'évaluation principal d'efficacité de l'étude CA209743 était la SG. Les autres critères d'efficacité étaient la SSP, l'ORR, et la durée de réponse, évalués par un comité de revue indépendant centralisé en aveugle (Blinded Independent Central Review BICR), en utilisant les critères RECIST modifiés.
L'étude a démontré une amélioration statistiquement significative en SG pour les patients randomisés dans le bras ipilimumab en association au nivolumab comparé à la chimiothérapie, lors de l'analyse intermédiaire pré-spécifiée, après l'observation d'au moins 419 événements (89% du nombre prévu d'événements pour l'analyse finale). Le suivi minimum pour la SG était de 22 mois.
Les résultats d'efficacité sont présentés dans la figure 1 et dans le Tableau 4.
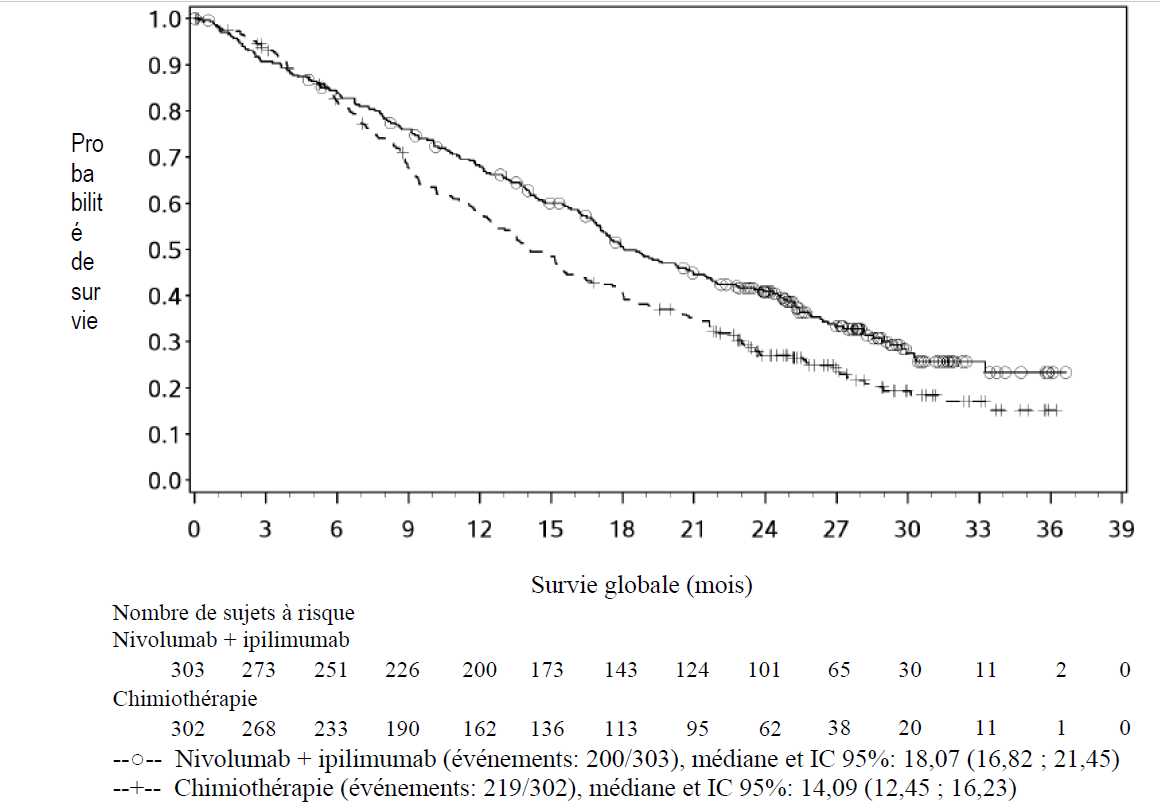
Tableau 4: Résultats d'efficacité (CA209743)
| | ipilimumab + nivolumab (n = 303) | chimiothérapie (n = 302) |
| Survie Globale Evénements | 200 (66%) | 219 (73%) |
| Hazard ratio | 0,74 | |
| (96.6% CI)b | (0,60 ; 0,91) | |
| Log-rank stratifié p-valuec | 0,002 | |
| Médiane (mois)a | 18,1 | 14,1 |
| (IC 95%) | (16,8 ; 21,5) | (12,5 ; 16,2) |
| Taux (95% CI) à 24 moisa | 41% (35,1 ; 46,5) | 27% (21,9 ; 32,4) |
| Survie sans progression Evénements | 218 (72%) | 209 (69%) |
| Hazard ratio | 1,0 | |
| (IC 95%) b | (0,82 ; 1,21) | |
| Log-rank stratifié p-valuec | 0,0001 | |
| Médiane (mois)a (IC 95%) | 6,8 (5,6 ; 7,4) | 7,2 (6,9 ; 8,1) |
| Taux de réponse objective | 40% | 43% |
| (IC 95%) | (34,1 ; 45,4) | (37,1 ; 48,5) |
| Réponse complète (RC) | 1,7% | 0 |
| Réponse partielle (RP) | 38% | 43% |
| Durée de réponse | | |
| Médiane (mois)a | 11,0 | 6,7 |
| (IC 95%) | (8,1 ; 16,5) | (5,3 ; 7,1) |
| % avec durée ≥ 6 mois | 69% | 53% |
a Estimation de Kaplan-Meier.
b Modèle à risques proportionnels de Cox stratifié.
c La valeur p est comparée à l'alpha attribué de 0,0345 pour cette analyse intermédiaire
44,2% et 40,7% des patients ont reçu un traitement systémique ultérieur dans les bras en association et chimiothérapie, respectivement. Une immunothérapie ultérieure (incluant anti-PD-1, anti-PD-L1, et anti-CTLA-4) a été reçue par 3,3% et 20,2% des patients dans les bras en association et chimiothérapie, respectivement.
Les résultats de SG chez les patients avec une expression tumorale de PD-L1 < 1% (HR [IC 95%] étaient de 0,94 [0,62 ; 1,40], n = 57) et de 0,69 [0,55 ; 0,87], n = 232) chez les patients avec une expression tumorale de PD-L1 ≥1% (HR [IC 95%].
Le tableau 5 résume les résultats d'efficacité pour la SG, la SSP et l'ORR par histologie dans les analyses pré-spécifiées en sous-groupes.
Tableau 5 : Résultats d'efficacité par histologie (CA209743)
| Epithélioïde (n = 471) | Non-épithélioïde (n = 134) | |||
| | ipilimimab | chimiothérapie | ipilimumab | chimiothérapie |
| | + | (n = 235) | + | (n = 67) |
| | nivolumab | | nivolumab | |
| | (n = 236) | | (n = 67) | |
| Survie globale | | | | |
| Evénements | 157 | 164 | 43 | 55 |
| Hazard ratio (95% CI)a | 0,85 (0,68; 1,06) | 0,46 (0,31 ; 0,70) | ||
| Médiane (mois) | 18,73 | 16,23 | 16,89 | 8,80 |
| (IC 95%) | (17,05 ; 21,72) | (14,09 ; 19,15) | (11,83 ; 25,20) | (7,62 ; 11,76) |
| Taux (95% CI) à 24 moisa | 41,2 (34,7 ; 47,6) | 31,8 (25,7 ; 38,1) | 39,5 (27,5 ; 51,2) | 9,7 (3,8; 18,9) |
| Survie sans progression | | | | |
| Hazard ratio (IC 95%)a | 1,14 (0,92; 1,41) | 0,58 (0,38 ; 0,90) | ||
| Taux de réponse objective | 38,6% | 47,2% | 43,3% | 26,9% |
| (IC 95%) b | (32,3 ; 45,1) | (40,7 ; 53,8) | (31,2 ; 56,0) | (16,8 ; 39,1) |
| Durée de réponse | 8,44 | 6,83 | 24,02 | 4,21 |
| Médiane (mois) | (7,16 ; 14,59) | (5,59 ; 7,13) | (8,31 ; N.A.) | (2,79 ; 7,03) |
| (IC 95%)c | ||||
a Rapport des risques basé sur le modèle à risques proportionnels de Cox non stratifié
b Intervalle de confiance basé sur la méthode de Clopper et Pearson
c Médiane calculée selon la méthode de Kaplan-Meier
Un total de 157 patients atteints de MPM et âgés de ≥ 75 ans ont été inclus dans l'étude CA209743 (78 dans le bras ipilimumab en association au nivolumab et 79 dans le bras chimiothérapie). Un HR de 1,02 (IC 95% : 0,70 ; 1,48) en SG a été observé pour ipilimumab en association au nivolumab vs. chimiothérapie au sein de ce sous-groupe d'étude. Un taux plus élevé d'effets indésirables graves et d'arrêt de traitement dû à des effets indésirables a été montré chez les patients âgés de 75 ans ou plus comparé à l'ensemble des patients ayant reçu ipilimumab en association au nivolumab (voir rubrique Effets indésirables). Cependant, compte tenu de la nature exploratoire de cette analyse en sous-groupe, aucune conclusion définitive ne peut être tirée.
La pharmacocinétique d'ipilimumab a été étudiée chez 785 patients atteints de mélanome avancé ayant reçu 4 doses d'induction allant de 0,3 à 10 mg/kg administrées une fois toutes les 3 semaines. Les Cmax, Cmin et ASC d'ipilimumab étaient proportionnelles à la dose, dans la fourchette de doses étudiée. Après dosages répétés d'ipilimumab administré toutes les 3 semaines, la clairance est restée constante, et une accumulation systémique minime a été observée comme en témoigne un indice d'accumulation de 1,5 fois ou moins. L'état d'équilibre d'ipilimumab a été atteint après la troisième injection. Dans une analyse pharmacocinétique de population, les paramètres de moyenne (pourcentage du coefficient de variation) suivants d'ipilimumab ont été observés : demi-vie de 15,4 jours (34,4%); clairance systémique de 16,8 ml/h (38,1%); et volume de distribution à l'état d'équilibre de 7,47 L (10,1%). La moyenne (pourcentage du coefficient de variation) de Cmin d'ipilimumab à l'état d'équilibre pour un traitement d'induction à 3 mg/kg était de 19,4 µg/ml (76,6%).
La clairance d'ipilimumab était augmentée lorsque le poids et le taux de LDH à l'inclusion étaient plus élevés. Cependant, aucune adaptation posologique n'est nécessaire selon les taux de LDH et le poids après administration en mg/kg. La clairance n'a pas été modifiée par l'âge (entre 23 et 88 ans), le sexe, l'utilisation concomitante de budésonide ou de dacarbazine, l'état général, le statut HLA-A2*0201, l'insuffisance hépatique légère, l'insuffisance rénale, l'immunogénicité et les traitements anticancéreux antérieurs. Les effets liés à la race n'ont pas été étudiés car les données provenant de groupes éthniques non-Caucasiens étaient insuffisantes. Aucune étude pharmacocinétique n'a été conduite pour évaluer la pharmacocinétique d'ipilimumab dans la population pédiatrique ou chez les patients présentant une insuffisance rénale ou hépatique.
Dans une analyse dose réponse chez 497 patients atteints d'un mélanome avancé, la SG s'est montrée indépendante des traitements anticancéreux systémiques antérieurs et augmentée avec des concentrations plasmatiques d'ipilimumab Cminss plus élevées.
Yervoy en association à nivolumab: Lorsque ipilimumab 1 mg/kg était administré en association à nivolumab 3 mg/kg, la clairance d'ipilimumab a diminué de 1,5% et la clairance de nivolumab a été augmentée de 1%, ce qui n'a pas été considéré comme cliniquement pertinent.
Lorsqu'il est administré en association avec le nivolumab, la clairance d'ipilimumab est augmentée de 5,7% en présence d'anticorps anti-ipilimumab et la clairance de nivolumab est augmentée de 20% en présence d'anticorps anti-nivolumab. Ces changements n'ont pas été considérés cliniquement pertinents.
Insuffisance rénale
Dans l'analyse pharmacocinétique de population des données d'études cliniques chez des patients atteints de mélanome métastatique, la présence d'une insuffisance rénale préexistante légère et modérée n'a pas influencé la clairance d'ipilimumab. Les données cliniques et pharmacocinétiques avec une insuffisance rénale sévère préexistante sont limitées; la nécessité d'un potentiel ajustement de dose ne peut être déterminée.
Insuffisance hépatique
Dans l'analyse pharmacocinétique de population des données d'études cliniques chez des patients atteints de mélanome métastatique, la présence d'une insuffisance hépatique préexistante légère n'a pas influencé la clairance d'ipilimumab. Les données cliniques et pharmacocinétiques avec une insuffisance hépatique modérée préexistante sont limitées; la nécessité d'un ajustement de dose ne peut être déterminée. Aucun patient atteint d'insuffisance hépatique sévère préexistante n'a été identifié dans les études cliniques.
YERVOY a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines.
Du fait des effets indésirables possibles tels que la fatigue (voir rubrique Effets indésirables), les patients doivent être avisé d'être prudents lorsqu'ils conduisent ou utilisent des machines tant qu'ils ne sont pas certains qu'ipilimumab n'altère pas leur vigilance.
Au cours des études d'administration IV répétée chez le singe, ipilimumab était généralement bien toléré. Des effets indésirables d'origine immunologique étaient peu fréquemment observés (environ 3%) et comprenaient colites (dont un seul cas s'est avéré fatal), dermatites, et réactions liées à la perfusion (possiblement dues à la libération massive de cytokines consécutive à une vitesse de perfusion trop rapide). Une diminution de la masse de la thyroïde et des testicules a été observée au cours d'une étude sans être accompagnée d'observations histopathologiques ; la pertinence clinique de ce constat n'est pas connue.
Les effets de l'ipilimumab sur le développement pré et post-natal ont été étudiés dans une étude réalisée chez des singes Cynomolgus. Les singes gravides recevaient ipilimumab toutes les 3 semaines du début de l'organogénèse durant le premier trimestre jusqu'à la délivrance, à des niveaux d'exposition (ASC) similaires ou supérieurs à ceux associés à la dose recommandée de 3 mg/kg d'ipilimumab. Aucun effet indésirable lié au traitement sur la reproduction n'a été détecté pendant les deux premiers trimestres de la grossesse. A partir du troisième trimestre, les 2 groupes ipilimumab ont présenté des incidences plus élevées d'avortement, de mortalité néonatale, d'accouchement prématuré (avec hypotrophie correspondante) et de mortalité infantile par rapport aux animaux contrôles ; ces résultats étaient dose-dépendants. De plus, 2 nouveau-nés exposés in utero à l'ipilimumab ont présenté des anomalies du développement externe ou des anomalies viscérales au niveau du système génito- urinaire. L'un de sexe féminin a présenté une agénésie rénale unilatérale du rein gauche et de l'uretère, et l'autre de sexe masculin présentait une imperforation de l'urètre avec une obstruction urinaire associée et un oedème scrotal sous-cutané. La relation entre ces malformations et le traitement n'est pas claire.
Des études pour évaluer le potentiel mutagène et carcinogène d'ipilimumab n'ont pas été menées. Des études de fertilité n'ont pas été menées.
La préparation doit être réalisée par du personnel formé conformément aux règles de bonnes pratiques, particulièrement en ce qui concerne le respect de l'asepsie.
Calculer la dose :
Ipilimumab en association à nivolumab :
La dose prescrite au patient est donnée en mg/kg. En fonction de cette dose prescrite, calculer la dose totale à administrer. Plus d'un flacon de solution à diluer de YERVOY peut être nécessaire pour obtenir la dose totale pour le patient.
§ Chaque flacon de 10 ml de solution à diluer de YERVOY donne 50 mg d'ipilimumab.
§ Dose totale d'ipilimumab en mg = poids du patient en kg × la dose prescrite en mg/kg.
§ Volume de solution à diluer de YERVOY pour préparer la dose (ml) = dose totale en mg divisée par 5 (la concentration de la solution à diluer de YERVOY est de 5 mg/ml).
Préparer la perfusion :
S'assurer d'opérer dans des conditions aseptiques lorsque vous préparez la perfusion.
YERVOY peut être utilisé par administration intraveineuse soit :
§ sans dilution, après transfert dans un récipient pour perfusion en utilisant une seringue stérile appropriée
ou
§ après une dilution jusqu'à 5 fois le volume initial de solution à diluer (jusqu'à 4 parties de diluant pour 1 partie de solution concentrée). La concentration finale sera comprise entre 1 et 4 mg/ml. Pour diluer la solution à diluer de YERVOY, vous pouvez utiliser soit :
§ une solution injectable de chlorure de sodium à 9 mg/ml (0,9%) ; ou
§ une solution injectable de glucose à 50 mg/ml (5%)
ETAPE 1
§ Laisser la quantité nécessaire de flacons de YERVOY à température ambiante pendant approximativement 5 minutes.
§ Inspecter la solution à diluer de YERVOY pour mettre en évidence la présence de particules étrangères ou d'un changement de coloration. La solution à diluer de YERVOY est claire à légèrement opalescente, incolore à jaune pale pouvant contenir quelques particules. Ne pas utiliser si une quantité anormale de particules et si des signes de changement de coloration sont présents.
§ Retirer le volume nécessaire de solution à diluer de YERVOY en utilisant une seringue stérile appropriée.
ETAPE 2
§ Transférer la solution à diluer dans une bouteille en verre stérile et évacuée ou une poche pour perfusion intraveineuse (PVC ou non-PVC) sous vide.
§ Le cas échéant, diluer la solution concentrée avec le volume nécessaire de solution injectable de chlorure de sodium à 9 mg/ml (0,9%) ou de glucose à 50 mg/ml (5%). Afin de faciliter la préparation, la solution à diluer peut directement être transvasée dans une poche pré-remplie contenant le volume approprié de solution injectable de chlorure de sodium à 9 mg/mL (0,9%) ou de solution injectable de glucose à 50 mg/mL (5%). Mélanger doucement la perfusion par rotation manuelle.
Administration :
La perfusion de YERVOY ne doit pas être administrée en IVD ni en bolus IV. Administrer la perfusion de YERVOY en intraveineux sur une période de 30 minutes.
La solution de YERVOY ne doit pas être perfusée simultanément avec d'autres médicaments sur la même ligne intraveineuse. Utiliser une ligne intraveineuse séparée pour la perfusion.
Utiliser un set de perfusion et un filtre stérile, apyrogène, à faible liaison aux protéines (diamètre des pores de 0,2 µm à 1,2 µm).
La perfusion de YERVOY est compatible avec :
§ Les sets de perfusion en PVC.
§ Les filtres en ligne de polyethersulfone (0,2 µm à 1,2 µm) et de nylon (0,2 µm).
A la fin de la perfusion, rincer la ligne avec une solution injectable de chlorure de sodium à 9 mg/ml (0,9%) ou de glucose à 50 mg/ml (5%).
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Médicament réservé à l'usage hospitalier.
Prescription réservée aux spécialistes en oncologie ou aux médecins
compétents en cancérologie. Médicament nécessitant une surveillance
particulière pendant le traitement.
Solution à diluer pour perfusion (solution stérile).
Solution claire à légèrement opalescente, incolore à jaune pale qui peut contenir quelques particules, et a un pH de 7,0 et une osmolarité de 260-300 mOsm/kg.
10 ml de solution à diluer en flacon (verre de type I) muni d'un bouchon (caoutchouc butyl enrobé) et d'un opercule amovible (aluminium). Boîte de 1.
Chaque ml de solution contient 5 mg d'ipilimumab. Un flacon de 10 ml contient 50 mg d'ipilimumab.
Ipilimumab est un anticorps monoclonal entièrement humain anti-CTLA-4 (IgG1κ) produit dans des cellules ovariennes de hamster chinois par la technique de l'ADN recombinant.
Excipients à effet notoire :
Chaque ml de solution à diluer contient 0,1 mmol de sodium, soit 2,30 mg de sodium. Pour la liste complète des excipients, voir rubrique Liste des excipients.
Tris hydrochlorure (2-amino-2-hydroxymethyl-1,3-propanediol hydrochloride)
Chlorure de sodium
Mannitol (E421)
Acide pentétique (acide diéthylène-triamine-penta-acétique)
Polysorbate 80
Hydroxyde de sodium (pour ajuster le pH)
Acide chlorhydrique (pour ajuster le pH)
Eau pour préparations injectables